Research
We study the evolutionary processes that give rise to genetic and phenotypic differences between individuals, populations and closely related species. Specifically, we use mathematical models to better understand these processes, and statistical analyses to identify their footprints in data and make inferences about them. Our current work focuses primarily on the evolutionary causes of adaptation and disease, but we also study a variety of other topics.
Natural selection in the genome of humans and other species. Despite the pivotal role of natural selection in evolution, our knowledge about its genetic basis is still tentative. Fundamental questions, such as how many of the changes to the genome are beneficial, their typical selective advantage, and whether they arise from new mutations or from genetic variation that preceded changes in selection pressures, remain largely unanswered. One approach to learning about selective processes is based on learning to read the footprints that they leave in patterns of genetic variation. The deluge of genome-wide polymorphism and divergence data that has recently become available has therefore enabled rapid progress in this area.
To illustrate this approach, consider the case in which a new, beneficial mutation arises and rapidly sweeps to fixation in the population. As Maynard Smith and Haigh first described more than forty years ago (Maynard Smith & Haigh Genet Res 1974), a segment of the haplotype on which the beneficial mutation arose will be dragged (or hitchhike) to higher population frequency or fixation along with it. As a result, diversity levels will be reduced near beneficial substitutions that occurred recently (Fig. 1). These considerations imply that when we look at the average diversity levels as a function of distance from putatively functional substitutions, we should see a trough, if a subset of the substitutions were driven by selective sweeps. Moreover, our modeling shows that the depth of this trough reflects the fraction of selected substitutions and its width the strength of selection.

Figure 1: A selective sweep. The horizontal lines denote haplotypes in the population and white circles denote neutral mutant alleles at polymorphic sites in the genome. When a beneficial mutation (in green) fixes, it carries part of the haplotype on which it arose (in black) to high frequencies or fixation in the population, resulting in reduced genetic diversity near the selected site.
When we applied this approach to genome-wide polymorphism data from Drosophila, we found a striking signature of sweeps: a pronounced trough in diversity levels extending over ~15 kb around amino acid substitutions but not around silent (i.e., putatively neutral) ones, used as a control (Fig. 2A). By fitting a model to the trough, we inferred that most of the reduction in diversity was generated by a minority of strongly selected substitutions, while the remaining reduction was due to many more substitutions with much weaker effects.
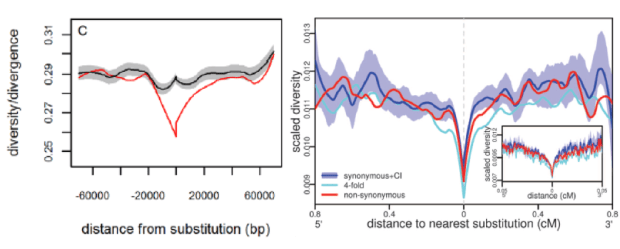
Figure 2: The footprints of selective sweeps in Drosophila (A) and their lack in humans (B). In Drosophila, silent sites were used to estimate average neutral diversity levels, whereas in humans, non-conserved, non-coding sites were. For details, see Sattath et al. (2011), Hernandez et al. (2011) and Elyashiv et al. (2016).
Interestingly, when we applied the same approach to humans (in collaboration with Molly Przeworski), it revealed an entirely different picture: diversity levels were no more reduced around amino acid substitutions than around silent ones. We further showed that the trough seen around both kinds of substitutions is in rough accordance with what is expected from background selection, i.e., the reduction in linked diversity levels caused by selection against deleterious mutations in or near genes (Charlesworth et al. Genetics 1993). Together with other lines of evidence (e.g., Coop et al. PLoS Genetics 2009), these results indicate that classic selective sweeps were rare in the past 250,000 years of human evolution and suggest that a change in focus to other modes of adaptation was warranted. A plausible alternative is that adaptation on the human lineage instead tended to involve polygenic changes to quantitative traits (Pritchard et al. Curr Biol 2010; see below).
This is one example of how our lab is using the recent deluge of genetic variation data to advance our understanding of natural selection in the genome. As our approaches improve, our predictions of patterns of genetic variation are becoming more precise (Fig. 3) and our inferences based on different footprints of natural selection are assembling into an increasingly coherent picture. This progress suggests that answers to enduring questions about the genetic basis of natural selection in the genome are perhaps now within reach.
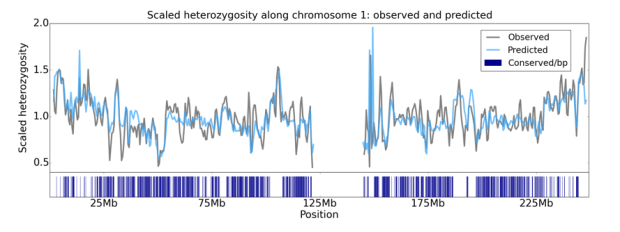
Figure 3: A comparison of observed and predicted diversity level along chromosome 1 of the human genome (Murphy et al., In preparation; see also McVicker et al. PLoS Genetics 2009). The footprints of background selection along the human genome were used to infer the strength and prevalence of purifying selection acting on different functional regions in the genome, such as coding and regulatory regions (the density of which is shown in blue with the vertical hashes). These parameters were then used to predict expected diversity levels in windows of 1 Mb (in blue), shown here alongside the observed levels in these windows (in gray).
Selected publications
Elyashiv, E., Sattath S., Hu, T., McVicker, G., Strutsovsky, A., Andolfatto, P., Coop, G. and G. Sella (2016). A genomic map of the effects of linked selection in Drosophila. PLoS Genetics 12: e1006130.
Hernandez, R. D., Kelley, J. L., Elyashiv, E., Melton, S. C., Auton, A., McVean, G., 1000 Genomes Project, Sella G.+ and M. Przeworski+ (2011) Classic selective sweeps were rare in recent human evolution. Science 331: 920-4. +Joint senior authors.
Sattath, S., Elyavish, E., Kolodny, O., Rinott, Y., and G. Sella (2011) Pervasive adaptive protein evolution apparent from diversity patterns around amino-acid substitutions in Drosophila simulans. PLoS Genetics 7: e1001302.
Elyavish, E., Bullaughey, K., Sattath, S., Rinott, Y., Przeworski, M. and G. Sella (2010) Variation in the intensity of purifying selection: an analysis of genome-wide polymorphism data from two closely related yeast species. Genome Research 20: 1558-73.
Sella, G., D. A. Petrov, M. Przeworski, and P. Andolfatto (2009) Evidence for pervasive natural selection in Drosophila? PLoS Genetics 5: e1000495.
M. Macpherson*, G. Sella*, J. C. Davis, and D. A. Petrov (2007) Genome-wide spatial correspondence between non-synonymous divergence and neutral polymorphism reveals extensive adaptation in Drosophila. Genetics 177: 2083-89. *Joint first authors.
The population genetics of quantitative genetic variation. Many phenotypes that we care about are “quantitative”, meaning that heritable variation in the traits is due to small contributions from many genetic variants segregating in the population. Quantitative phenotypes include morphological and life history traits (e.g., height, body mass index, and age of menarche) as well as the risk of developing many diseases (e.g., diabetes, MS, and schizophrenia). Although they have been studied for over a century, it only recently became possible to systematically dissect their genetic basis. Notably, over the past decade, Genome Wide Association Studies (GWASs) in humans have identified thousands of variants reproducibly associated with hundreds of quantitative traits, including the susceptibility to a wide variety of diseases. These studies are beginning to reveal the genetic architecture of quantitative traits (e.g., the number of variants underlying genetic variation, alongside their effect sizes and frequencies), as well as intriguing differences in architecture among traits.
Our work aims to ground the findings from human GWASs in a better understanding of the processes that give rise to quantitative genetic variation. In so doing, we hope to address long-standing questions about how quantitative genetic variation is maintained in natural populations and why it differs among different kinds of traits (e.g., morphological and life history traits or early and late onset diseases). From a practical perspective, we hope to learn about the underpinnings of “missing heritability”, and thus inform the design of mapping studies and phenotypic prediction efforts.
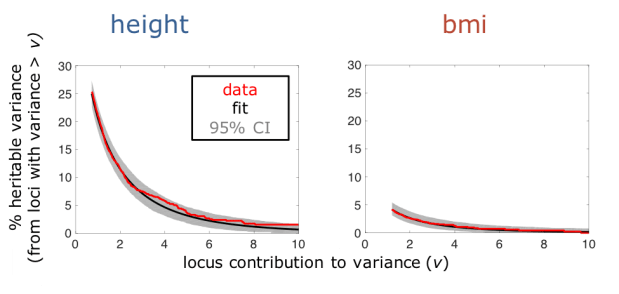
Figure 4: Comparing theoretical predictions and observations from GWAS for human height and body mass index (bmi). The graphs show the distribution of genetic variance among loci, with the contribution of a locus to variance on the x-axis and the proportion of the variance coming from loci that contribute that much or more on the y-axis. Despite considerable data for both traits, our modeling predictions based on only two parameters cannot be rejected (Kolmogorov-Smirnov p=0.22 for height and 0.67 for bmi)—in other words, our model provides a good fit to the data.
We are also interested in how quantitative traits respond to changing selection pressures. As some modern human populations first migrated from Africa to the colder climate of Europe, for example, we might imagine that there would be selection for a change in body size, in order to increase the surface-to-volume ratio and minimize heat dissipation (i.e., following the Bergman-Allen rule). Assuming, as seems highly plausible, that this trait is quantitative, we would therefore expect a polygenic adaptive response, involving changes to allele frequencies at many loci that affect the trait and were segregating in the population before the novel selection pressure (see above). We currently do not understand quite what these changes in allele frequencies should look like or how to find them in variation data. To obtain these answers, our work marries modeling approaches from evolutionary genetics and findings from human genetics.
Selected publications
Heyward, L. and G. Sella (2019). Polygenic adaptation after a sudden change in environment. BioRxiv 792952.
Sella, G. and N. Barton (2019). Thinking about the evolution of complex traits in the era of GWAS. Annual Reviews in Genomics and Human Genetics 20: 461-493.
Simons, Y., Bullaughey, K., Hudson, R. and G. Sella (2018). A population genetic interpretation of GWAS findings for human quantitative traits. PLoS Biol 16: e2002985.
Simons, Y. and G. Sella (2016). The impact of recent population history on the deleterious mutation load in humans and close evolutionary relatives. Curr Opin Genet Dev 41: 150-158.
Simons, Y., Turchin, M., Pritchard, J. K.+, and G. Sella+ (2014). The deleterious mutation load is insensitive to recent population history. Nature Genetics 46: 220–224. +Joint senior authors.
